
Consent Form Templates
- ICF template - exempt studies ( updated 11/27/24 )
- ICF template - full board and expedited studies ( updated 11/27/24 )
- HIPAA Language for Studies Involving Protected Health Information
- GINA Language for Studies Involving Genetic Analysis
- Assent Form: 6-12 Years
- Assent Form: 13-17 Years
What is Informed Consent?
Informed consent for research participation is a fundamental part of protecting human research subjects. This idea is at the core of one of the Belmont Report’s ethical principles, respect for persons. The federal regulations governing research with human subjects contain criteria for what information must be provided to potential research participants before they decide whether to be part of a research study, as well as specific criteria for when an IRB can waive some or all of the informed consent requirements.
Informed consent is a process. More than just a document, the informed consent process must:
- provide all of the required information (known as “elements of consent”)
- allow the potential research subject adequate opportunity to review the information and time to make a decision
- afford them the opportunity to ask questions. The informed consent process should be designed in such a way to avoid pressure or coercion and maximize an individual's confidentiality
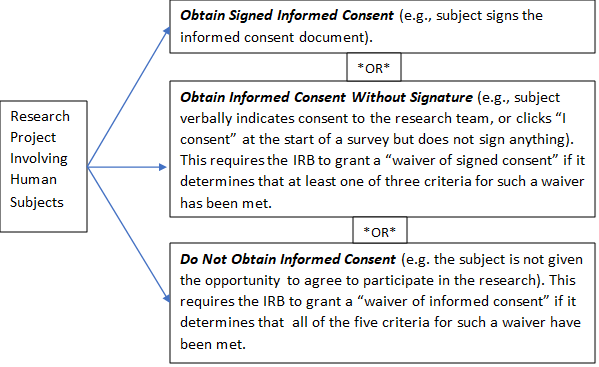
There are three options for informed consent:
Jump to a Specific Section:
- Required Elements of Informed Consent
- Informed Consent for Exempt Research
- Criteria for a Waiver of Signed Consent
- Criteria for a Waiver of Consent
- Criteria for an Alteration of Consent
- Informed Consent Documents in Languages other than English
- Impaired Capacity and Consent
- Child Assent
- Parental Permission
- Passive Consent/Passive Permission
Required Elements of Informed Consent
As described in the federal regulations (45 CFR 46 116 (b) and (c)), the following information must be provided to a potential research subject to allow them to make an informed decision as to whether or not to participate in a study that does not fit into an exempt category:
(1) A statement that the study involves research, an explanation of the purposes of the research and the expected duration of the subject’s participation, a description of the procedures to be followed, and identification of any procedures that are experimental;
(2) A description of any reasonably foreseeable risks or discomforts to the subject;
(3) A description of any benefits to the subject or to others that may reasonably be expected from the research;
(4) A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject;
(5) A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained;
(6) For research involving more than minimal risk, an explanation as to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained;
(7) An explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights, and whom to contact in the event of a research-related injury to the subject;
(8) A statement that participation is voluntary, refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is otherwise entitled; and
(9) One of the following statements about any research that involves the collection of identifiable private information or identifiable biospecimens:
(i) A statement that identifiers might be removed from the identifiable private information or identifiable biospecimens and that, after such removal, the information or biospecimens could be used for future research studies or distributed to another investigator for future research studies without additional informed consent from the subject or the legally authorized representative, if this might be a possibility; or
(ii) A statement that the subject’s information or biospecimens collected as part of the research, even if identifiers are removed, will not be used or distributed for future research studies.
The following additional elements of informed consent are required when applicable to the particular research study:
(10) A statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant) that are currently unforeseeable;
(11) Anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s or the legally authorized representative’s consent;
(12) Any additional costs to the subject that may result from participation in the research;
(13) The consequences of a subject’s decision to withdraw from the research and procedures for orderly termination of participation by the subject;
(14) A statement that significant new findings developed during the course of the research that may relate to the subject’s willingness to continue participation will be provided to the subject;
(15) The approximate number of subjects involved in the study;
(16) A statement that the subject’s biospecimens (even if identifiers are removed) may be used for commercial profit and whether the subject will or will not share in this commercial profit;
(17) A statement regarding whether clinically relevant research results, including individual research results, will be disclosed to subjects, and if so, under what conditions; and
(18) For research involving biospecimens, whether the research will (if known) or might include whole genome sequencing (i.e., sequencing of a human germline or somatic specimen with the intent to generate the genome or exome sequence of that specimen).
The informed consent templates provided by the Office of Research Compliance include all the required information noted above.
Informed Consent for Exempt Research
Informed consent is required even when the research meets the criteria for an exempt category, but the process and document can be shorter and simpler. Signed consent is usually not required for exempt research, unless there is an aspect of the research that does require a signature. A common example is use of FERPA-protected information in exempt category 1 research.
Criteria for a Waiver of Signed Consent
An IRB may waive the requirement for the investigator to obtain a signed informed consent form for some or all subjects if it finds any of the following:
1. That the only record linking the subject and the research would be the informed consent form and the principal risk would be potential harm resulting from a breach of confidentiality. Each subject (or legally authorized representative) will be asked whether the subject wants documentation linking the subject with the research, and the subject’s wishes will govern;
2. That the research presents no more than minimal risk of harm to subjects and involves no procedures for which written consent is normally required outside of the research context;
3. That the subjects or legally authorized representatives are members of a distinct cultural group or community in which signing forms is not the norm, that the research presents no more than minimal risk of harm to subjects and there is an appropriate alternative mechanism for documenting that informed consent was obtained.
If a waiver of signature is granted, the rest of the informed consent process is still required. If researchers are requesting a waiver of signed consent, the justification must be explained in the IRB submission.
Criteria for a Waiver of Informed Consent
In order to grant a waiver of the requirement to obtain informed consent for research, the IRB must find and document all of the following:
1. The research involves no more than minimal risk;
2. The research could not practicably be carried out without the waiver or alteration;
3. If the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format;
4. The waiver or alteration will not adversely affect the rights and welfare of the subjects;
5. Whenever appropriate, the subjects or legally authorized representatives will be provided with additional pertinent information after participation.
Even if the IRB grants a waiver of informed consent, an information sheet may be required. If researchers are requesting a full waiver of consent, the justification must be explained in the IRB application.
Criteria for an Alteration of Consent
An IRB may approve a consent procedure that omits some, or alters some or all, of the elements of informed consent. The most common reason for an alteration of consent is when the research involves deception, and the study design requires that participants are misled about some key aspect of the research or are provided with false information. In order to grant an alteration of consent, the IRB must find and document all of the following:
1. The research involves no more than minimal risk;
2. The research could not practicably be carried out without the waiver or alteration;
3. If the research involves using identifiable private information or identifiable biospecimens, the research could not practicably be carried out without using such information or biospecimens in an identifiable format;
4. The waiver or alteration will not adversely affect the rights and welfare of the subjects;
5. Whenever appropriate, the subjects or legally authorized representatives will be provided with additional pertinent information after participation.
Informed Consent Documents in Languages other than English
If you will be conducting study procedures with research participants in languages other than English, you will need to provide a copy of all translated documents, including the informed consent form, in the language you will be using with study participants for IRB review. You will also need to provide a Verification of Translation form to attest to the accuracy of the translated documents. This form may be signed by a member of the research team, a professional translation service, or another qualified individual. In certain cases, the IRB may require independent verification from someone outside the research team.
Impaired Capacity and Consent
Although adults are presumed to possess legal capacity to consent, some adults might have impaired or diminished capacity to consent to a specific research protocol due to health status or other factors. In order to provide voluntary informed consent, participants must be able to comprehend information, deliberate on choices offered in light of personal values, understand the consequences of consent (or refusal), and communicate a decision. Ethically, we must ensure that prospective research subjects who have, or might have, a diminished capacity to provide informed consent are offered opportunities to participate in research when possible, and that their assent is obtained in a way that is respectful and legally valid.
Investigators should assess consent capacity on an individual level, rather than judge capacity merely on the basis of an individual’s status (e.g., age, disability) or medical diagnosis. Assessment might involve informal interview techniques, validated assessment tools, or other strategies tailored to the specific research protocol. Re-assessment during the course of the study may be necessary to assure that participants are protected.
The federal regulations define a Legally Authorized Representative (LAR) as an individual or judicial or other body authorized under applicable law to consent on behalf of a prospective subject to the subject’s participation in the procedure(s) involved in the research. If there is no applicable law addressing this issue, an LAR is an individual recognized by institutional policy as acceptable for providing consent in the nonresearch context on behalf of the prospective subject to the subject’s participation the research. Researchers should carefully review written authorizations or appointments presented on behalf of a participant to determine whether they are broad enough to be used for research purposes. Ohio University recognizes the authority of an individual authorized by law to make medical decisions in the non-research context (e.g., plenary guardian; durable power of attorney for health care; statutory surrogate for health care) to consent to the same or similar types of medical procedures involved in the research. The IRB also may impose additional safeguards in the IRB-approved research protocol to protect the rights and welfare of the participant.
If it is determined that an individual lacks the cognitive capacity to consent to research participation, and the investigator decides to include an IRB-approved proxy decision maker (e.g. LAR) in the consent process, the investigator must obtain that individual’s permission and the prospective research subject’s assent, to the extent possible. The dissent of a prospective research subject must always be respected.
Child Assent
In the state of Ohio, a “child” is someone under the age of 18. Children are not able to consent to research, but whenever possible, they must be given the opportunity to indicate their assent to participate in a research study. In the federal regulations, assent is defined as a child's affirmative agreement to participate in research. Mere failure to object should not, absent affirmative agreement, be construed as assent. In determining whether children are capable of assenting, the IRB shall take into account the ages, maturity, and psychological state of the children involved. If the IRB determines that the children do not have the capability to assent, then under the federal regulations, the assent of the children is not a necessary condition for proceeding with the research. The IRB may also waive the requirement of child consent under the same criteria as a waiver of informed consent.
Parental Permission
In the federal regulations, parental permission is defined as the agreement of parent(s) or guardian(s) to have their child or ward participate in research. In most cases, researchers must obtain the permission of each child's parent(s) or guardian(s) before enrolling children into a research study. For research involving no more than minimal risk, or more than minimal risk but with the prospect of direct benefit to the child, the permission of one parent is usually sufficient. For all other research, the permission of both parents will be required.
The IRB may grant a waiver of parental permission using the same criteria for a waiver of consent, but this is uncommon and it is strongly preferred that parents be given the opportunity to give permission for their child to participate in research. If the IRB determines that a research protocol is designed for conditions or for a subject population for which parental or guardian permission is not a reasonable requirement to protect the subjects, it may waive the parental permission requirements, provided an appropriate mechanism for protecting the children is substituted. Ohio University has a strong preference for obtaining signed parental permission whenever feasible.
We suggest that you use the appropriate Informed Consent Form template and replace “you” with “your child” to create your parental permission form.
Passive Consent/Passive Permission
Though this phrase (also sometimes referred to as “implied consent”) is commonly used in research, the federal regulations do not reference this process. In reality, when the research participant or parent does not affirmatively indicate consent/permission, then consent/permission is not being obtained. It cannot be assumed that a failure to opt out equals consent to participate. Therefore, researchers seeking to use passive/implied consent/permission will need a waiver of informed consent/parental permission from the IRB.